【R】遺伝子発現解析を学んでみたい③
・お題:前回の続き。Differential expression analysis(DEG)という解析を少し齧ってみたい。私はWet専門で学んできた人間だから、この辺の解析方法にはとんと疎い。以下のHPを順になぞって学んでいきたいと思う。
・今回は前回の続きであり、データセットyから発現量の小さい遺伝子を除き、分布がサンプル間でそれほど大きく変わっていないことを確認した状態とする。参考サイトでいうと、Multidimensional scaling plotsの直前まで終わっている感じ。
・これから解析を始めていくわけだけれど、まずはどのサンプルとどのサンプルが似ているのか、をざっくり見てみたい。この時に利用されるのがMDSプロットというやつで、遺伝子発現量という超多次元データを主成分分析で次元削減して図示したプロットのことらしい。とりあえずやってみる。
・デフォルトだと、第一主成分と第二主成分とサンプル名のプロットがplotMDSというコマンドで作れる。
> plotMDS(y)
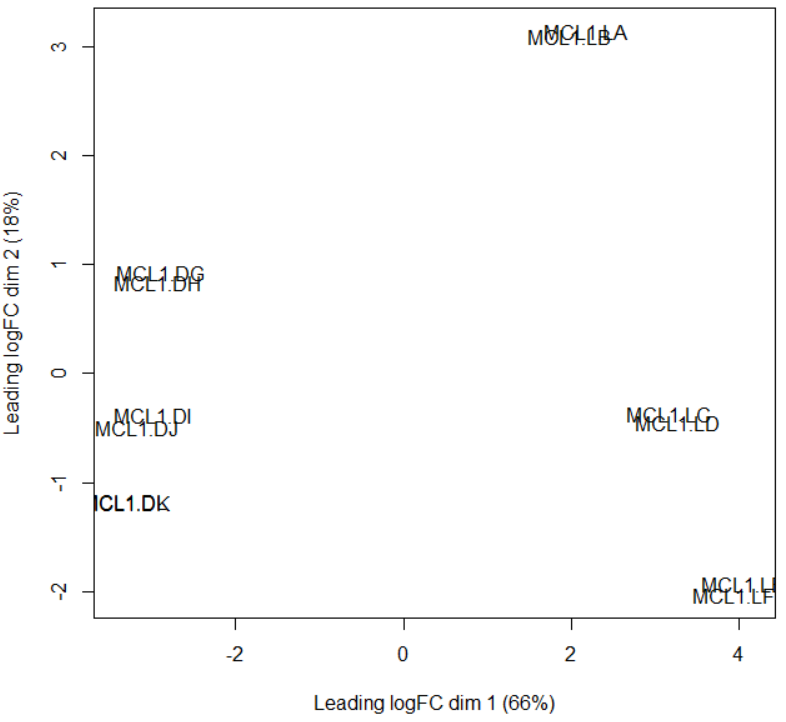
・なんだか、分かるような分からないような感じになっている。サンプル名が2つのペアとして配置されていることは分かるのだけれど、サンプル名ごとの実験条件がよく分からないので、解釈しづらい。全12サンプルの構成は、マウスの状態3種類×細胞2種類×2 replicatesになっているので、マウスの状態と細胞をそれぞれ図中に表現して、2 replicatesがどのように配置されているか見てみたい。
・サンプルの情報を確認する。CellTypeとStatusに細胞の種類とマウスの状態が格納されていることが分かる。
> sampleinfo
・細胞の種類ごとに図中の色を変えたい。col.celというオブジェクトを作成し、細胞の種類ごとにpurpleとorangeで色を格納したあと、col.celの色をもとにMDSプロットの色を変えたい。
> col.cell <- c("purple","orange")[sampleinfo$CellType]
> data.frame(sampleinfo$CellType,col.cell)

> plotMDS(y,col=col.cell)#色を指定してMDSプロットを作成
> legend("topleft",fill=c("purple","orange"),legend=levels(sampleinfo$CellType))#どの色が何の細胞か、レジェンドを加える。
> title("Cell type")
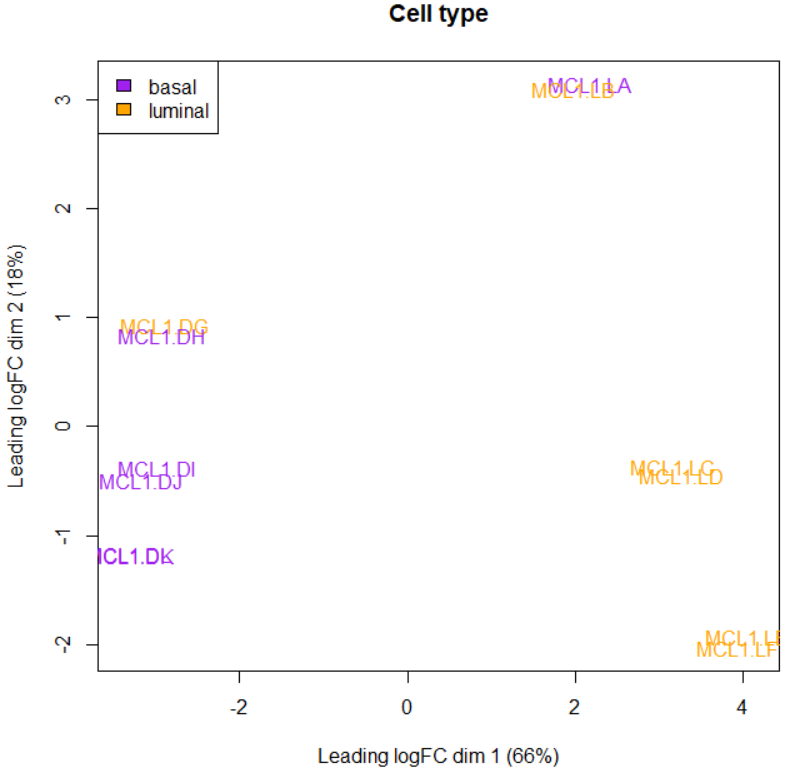
・一部のbasalとluminalのサンプルが被っていることが分かる。次にマウスの状態でも同じようにMDSプロットの色を変えてみる。
> col.status <- c("blue","red","black")[sampleinfo$Status]
> plotMDS(y,col=col.status)
> legend("topleft",fill=c("blue","red","black"),legend=levels(sampleinfo$Status),cex=0.8)
> title("Status")
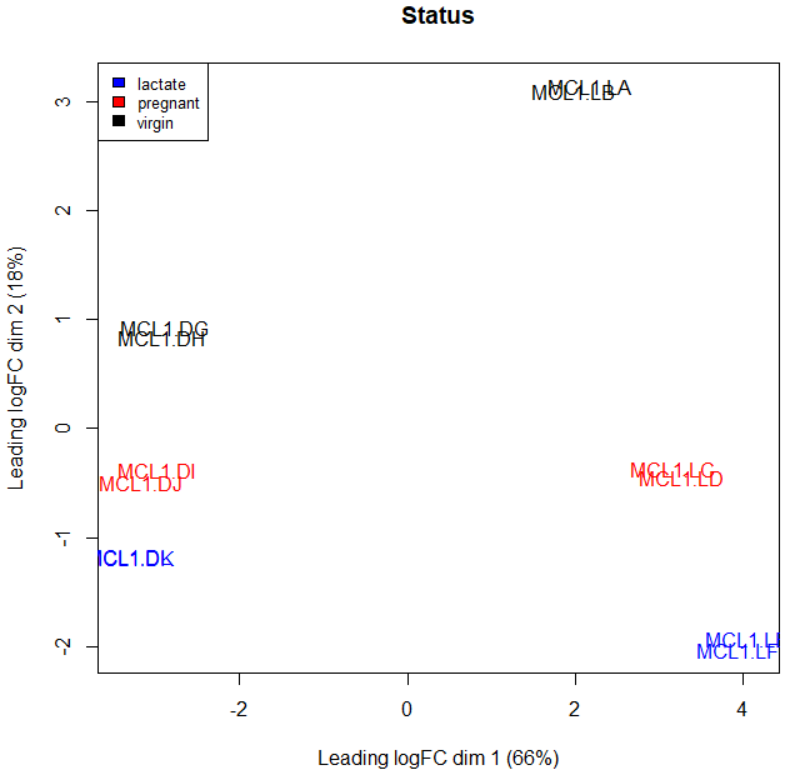
・こっちは混ざっていないぽい。どうやら、一部のサンプルでsampleinfoに誤りがあったらしい。元サイトの"SampleInfo_Corrected.txt"というファイルをサンプル情報として読み直す。
> sampleinfo <- read.delim("data/SampleInfo_Corrected.txt", stringsAsFactors = TRUE)
> group <- factor(paste(sampleinfo$CellType,sampleinfo$Status,sep="."))
> y$samples$group <- group
・これで図示してみる。せっかくなので、細胞の種類を色で、マウスの状態を形で表す。
> col.cell <- c("purple","orange")[sampleinfo$CellType]
> col.status <- c(1,2,3)[sampleinfo$Status]
> plotMDS(y,col=col.cell,pch = col.status)
> legend("topleft",fill=c("purple","orange"),legend=levels(sampleinfo$CellType))
> legend("topright",pch = c(1,2,3),legend=levels(sampleinfo$Status))
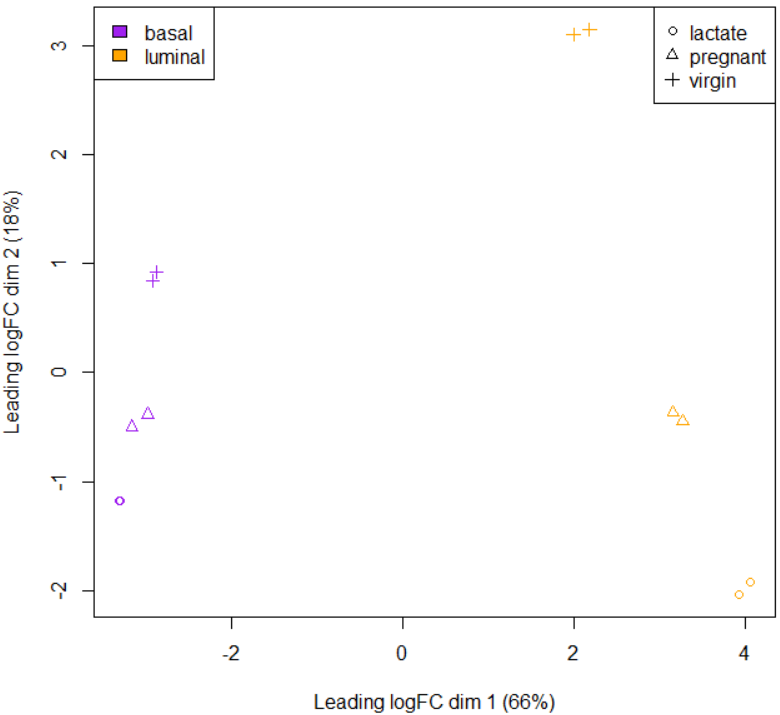
・綺麗に2つのサンプルがペアになって並んでおり、混ざっていない。第一主成分で細胞の種類が展開されており、左側にbasal、右側にluminalが配置されている。さらに、第二主成分ではマウスの状態が展開されており、上から順番にvirgin、pregnant、lactateが配置されている。
・他の主成分ではどのように配置されるか、見てみたい。GlimmaパッケージのglMDSPlot関数を使えば、インタラクティブなMDSプロットを作成できる。
> library(Glimma)
> labels <- paste(sampleinfo$SampleName, sampleinfo$CellType, sampleinfo$Status)
> glMDSPlot(y, labels=labels, groups=group, folder="mds")
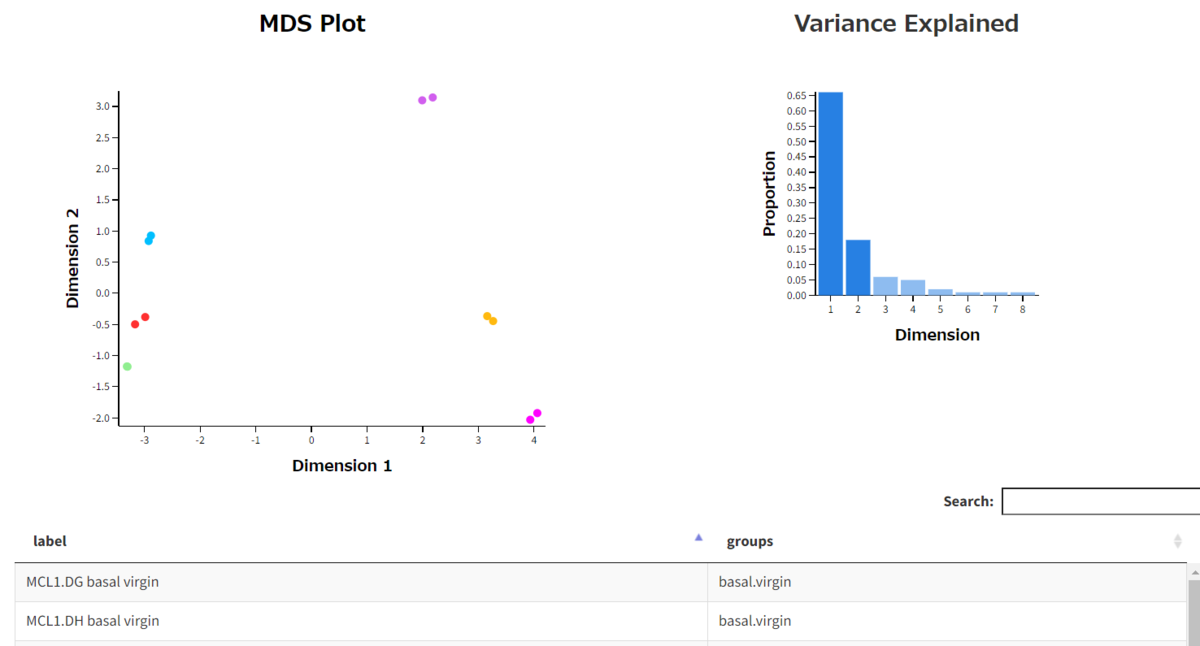
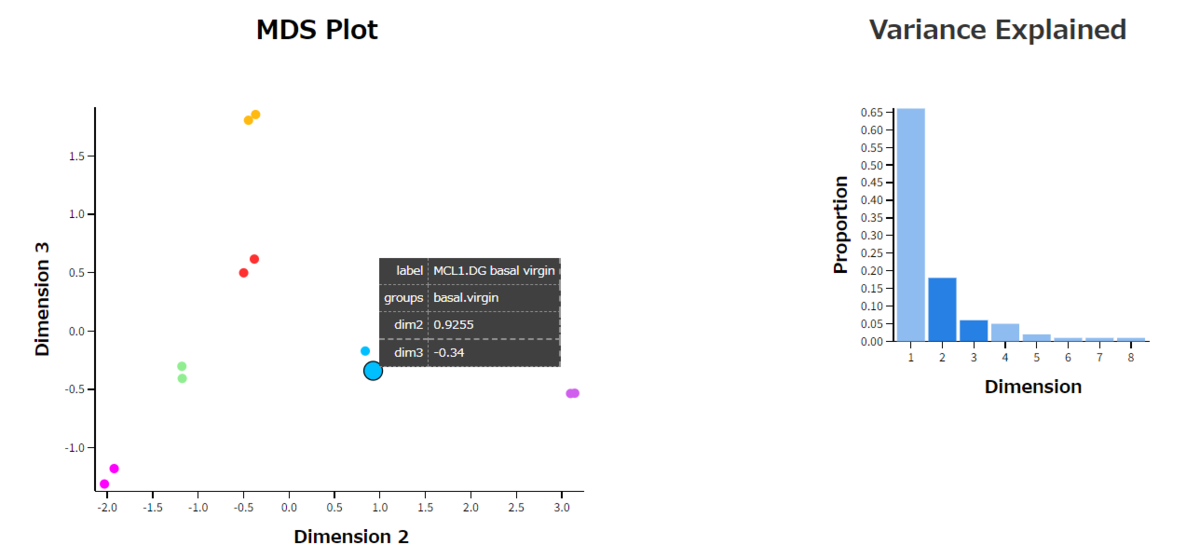
・例えば、第二主成分と第三主成分でMDSプロットを作成したければ、Variance Explainedの棒グラフをクリックすれば良い。MDS Platが自動で動く。さらに、カーソルをプロットに乗せるとサンプル情報がホバーする。
・次に、ヒートマップでサンプル間の遺伝子発現変動の傾向を覗いてみる。まず、変動の激しい遺伝子トップ500を抜き出す。
> var_genes <- apply(logcounts, 1, var)#各遺伝子の分散を算出。
> select_var <- names(sort(var_genes, decreasing=TRUE))[1:500]#分散の大きい遺伝子を降順に並べて、上位500個をとって来る。
> highly_variable_lcpm <- logcounts[select_var,]#該当する遺伝子の補正済み発現量を抜き出す。
> highly_variable_lcpm %>% head()

・変動の激しい遺伝子トップ500を使って、ヒートマップを描く。列は各サンプルを、行は各遺伝子を表す。列が何なのか分かりにくいので、細胞の種類で色分けしたバーを設定する(ColSideColors)。
> library(gplots)
> col.cell <- c("purple","orange")[sampleinfo$CellType]
> heatmap.2(highly_variable_lcpm,trace="none", main="Top 500 most variable genes across samples",ColSideColors=col.cell,scale="row")
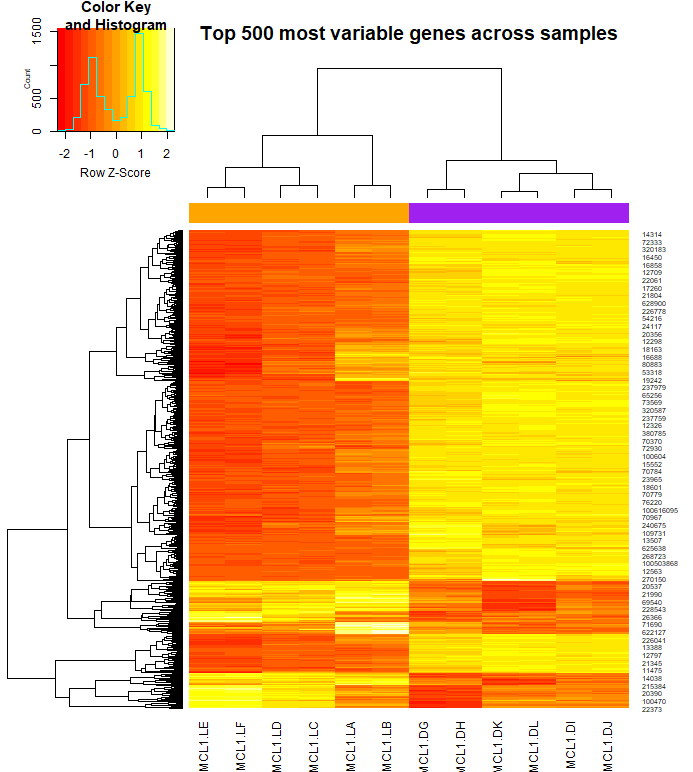
・なんだか色が分かりにくい。色をちょっと変えてみる。私の感覚的には、赤が高くて青が低く、変動がなければ色が薄い感じがそれっぽい(カラーユニバーシティ的にどうかはいったん置いておく)。
> heatmap.2(highly_variable_lcpm,trace="none", main="Top 500 most variable genes across samples",ColSideColors=col.cell,scale="row",col=bluered(256))
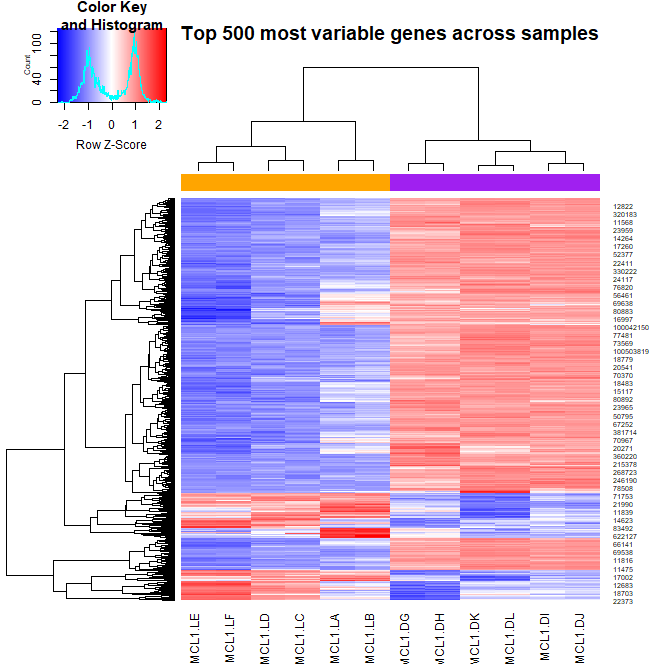
・列(サンプル)をもう少し細かく区別したい。heatmap.2ライブラリでは難しいので、別のライブラリを使う。NMFパッケージのaheatmap関数を使う。
> library(NMF)
> aheatmap(highly_variable_lcpm,col=bluered(256),main="Top 500 most variable genes across samples",annCol=sampleinfo[, 3:4],labCol=group, scale="row")
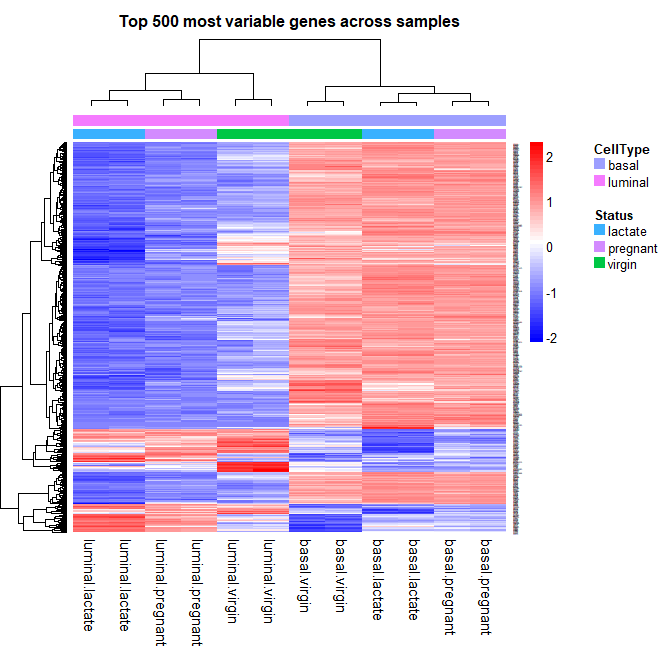
・どれがどのサンプルか、ちょっとわかりやすくなった。確かに、各サンプルで条件ごとに発現パターンが似ていそうな結果になった。階層型クラスタリングもそれっぽい感じになった。
つづく。