・お題:細胞一つ一つのオミクス情報を解析する手法をシングルセル解析と呼ぶらしい。Seuratというライブラリを使ってやってみたい。
・私は素人なので、以下のチュートリアルをなぞってみたい。
・インストールは以下を参照。
・チュートリアルの都合で、devtoolsとpatchworkもインストールする(割愛)。patchworkを使うと、別々の ggplot を同じグラフにうまいこと配置できるらしい。
・生データをチュートリアルのサイトからダウンロードして、カレントディレクトリに保存しておく。曰く、2,700個のPBMCの遺伝子発現データらしい。これは圧縮ファイルなので解凍する必要がある。Rのコマンドでうまく解凍できなかったので、7-zipで解凍した。.tar.gzファイルを解凍すると.tarファイルになり、.tarファイルを解凍すると「filtered_gene_bc_matrices」というフォルダができた。カレントディレクトリにこのフォルダを保存し、データを読み込んだ。
> library(tidyverse)
> library(Seurat)
> library(patchwork)
> pbmc.data <- Read10X(data.dir = "../filtered_gene_bc_matrices/hg19/")
・次に、Seuratオブジェクトを作成する。
> pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
Warning: Feature names cannot have underscores ('_'), replacing with dashes ('-')
> pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
・次に、品質の低い細胞を除く作業に入る。以下の方法論を参考にやるらしい。
・ミトコンドリア遺伝子が異様に高い細胞は死(にかけの)細胞なので、解析対象から除く。接頭辞MT-という遺伝子がミトコンドリア遺伝子であり、これを指標にミトコンドリア遺伝子を探し、異様に上がっている細胞を除くらしい。遺伝子名は以下のようになっている。ざっと出してみたけれど、全体の数が多すぎてMT-の遺伝子まで表示できない。。
> pbmc[ ["RNA"] ]@counts %>% row.names()

・PercentageFeatureSetで特定の機能セット(今回はミトコンドリア遺伝子)に属するすべてのカウントの割合を算出する。算出したカウントの割合はpercent.mtと名付ける。
> pbmc[ ["percent.mt"] ] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
> pbmc[ [ ] ] %>% head()
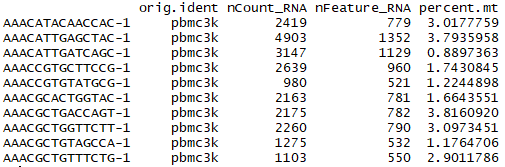
・2,700行(細胞数分)あり、各細胞におけるnFeature_RNA、nCount_RNAとpercent.mtが算出されている。nFeature_RNAは 検出された遺伝子の数、nCount_RNAはUMIの数、percent.mtはミトコンドリア遺伝子の割合を表す。UMIはUnique Molecular Identifiersのことで、その細胞中にあるmRNAの総数のことらしい。一つの遺伝子につき、複数のmRNAが転写されているであろうことから、nFeature_RNAはnCount_RNAよりも小さくなる。
・分布をバイオリンプロットで可視化してみる。
> VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)

・それぞれの関係を散布図で見てみる。
> plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
> plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
> plot1 + plot2

・クオリティの低い細胞を除き、良さそうなものを抽出する。ここでいう「クオリティが低い」は。nFeature_RNA(検出された遺伝子の数)が極端に少ない(<= 200)、極端に多い(>= 2500)、percent.mt(ミトコンドリア遺伝子の割合)が高い(>= 5)のものを意味するらしい。なぜ検出された遺伝子数が多いものも除くのかというと、複数の細胞がダマになってしまっている(正確には、ドロップレットに複数個の細胞が入ってしまっている)場合を除きたいかららしい。
> pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
・もう一度バイオリンプロットを描いてみると、以下のようになった(コードは割愛)。nFeature_RNAが高すぎる・低すぎるものと、percent.mtが高いものが確かに除かれたらしい。

・解析対象とする細胞の選別はここまでで、ここから遺伝子発現の解析をやっていく。
・まず、NormalizeDataという関数を使って、発現量をnormalizeする。関数中でnormalizeの手法とパラメータを指定しており、LogNormalizeという手法でnormalizeしているらしく、それぞれの遺伝子の発現量を遺伝子発現量の総量で割って10,000倍して対数化しているみたい。
> pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)#デフォルトのパラメータが記載の通りなので、この通りやるなら、パラメータをわざわざ指定しなくても良いぽい。
・細胞間で発現の変動が激しい遺伝子を調べてみる。FindVariableFeaturesというメソッドを使う。nfeaturesでVariableFeaturesを何個にするか設定する。今回は2000にして、Varianceが大きい順にトップ2000の遺伝子をVariableFeatures(発言の変動が激しい遺伝子)にするみたい。
> pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
> top10 <- head(VariableFeatures(pbmc), 10)
> top10
[1] "PPBP" "LYZ" "S100A9" "IGLL5" "GNLY" "FTL" "PF4" "FTH1" "GNG11" "S100A8"
・各遺伝子ごとの発現と分散の関係を、VariableFeaturePlot関数で図示する。
> plot1 <- VariableFeaturePlot(pbmc)#プロットする。
> plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)#プロットにラベルを付け加える。
> plot1 + plot2

・この後に次元削減をするけれど、スケールが揃っていないと次元削減を適用するのは難しい。各遺伝子の発現量をスケーリングして、すべての細胞での発現の平均が0に、細胞間の分散が1になるように、ScaleData関数を用いてスケーリングする。
> all.genes <- rownames(pbmc)
> pbmc <- ScaleData(pbmc, features = all.genes)
・次にPCAする。各主成分に対する各変数(遺伝子)の固有ベクトルの符号ごとの絶対値大きかったものも出てくる(各ベスト30個ずつの遺伝子が表示されている??)。
> pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))

> VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")#各主成分に対する各遺伝子の固有ベクトルが並ぶみたい。

> DimPlot(pbmc, reduction = "pca")#第1主成分と第2主成分で次元圧縮して各細胞をプロットした感じ。

・各主成分での各細胞における各遺伝子の主成分得点を見るのに、ヒートマップを描いてみる。細胞数を500個に指定しているので、各遺伝子ごとに500個のセルが並んでいる。
> DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)

・なんだかよく分からないので、以下を参考に色を変えてカラースケールバーを付けた。
> DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE, fast = FALSE)+
+ ggplot2::scale_fill_gradientn(colors = c("steelblue1", "white", "tomato"))

・なんとなく挙動の似ているor全然違う遺伝子群があるような気がする。
つづく。